To provide the best experiences, we use technologies like cookies to store and/or access device information. Consenting to these technologies will allow us to process data such as browsing behavior or unique IDs on this site. Not consenting or withdrawing consent, may adversely affect certain features and functions.
The technical storage or access is strictly necessary for the legitimate purpose of enabling the use of a specific service explicitly requested by the subscriber or user, or for the sole purpose of carrying out the transmission of a communication over an electronic communications network.
The technical storage or access is necessary for the legitimate purpose of storing preferences that are not requested by the subscriber or user.
The technical storage or access that is used exclusively for statistical purposes.
The technical storage or access that is used exclusively for anonymous statistical purposes. Without a subpoena, voluntary compliance on the part of your Internet Service Provider, or additional records from a third party, information stored or retrieved for this purpose alone cannot usually be used to identify you.
The technical storage or access is required to create user profiles to send advertising, or to track the user on a website or across several websites for similar marketing purposes.


This extends the material on an earlier thread “Analytical issues in causal diagrams: Barr art chart, Lombardi diagrams, evolutionary trees, Feynman diagrams, and some timelines” here.
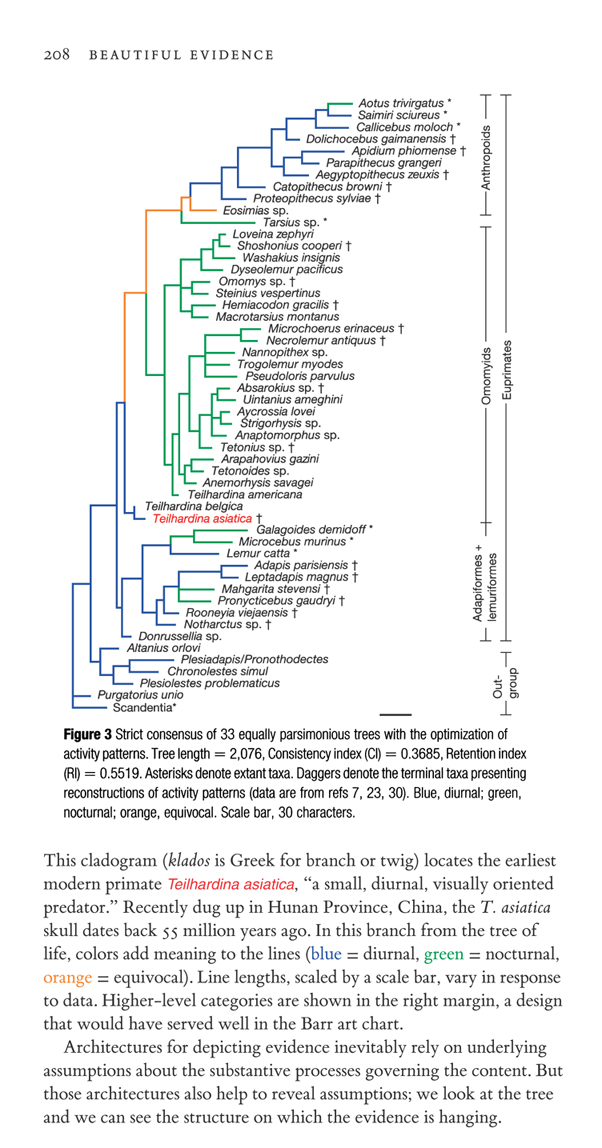
In an earlier post on Barr diagrams, ET asked what causal influences can be inferred from links or arrows in a graphic, and extended his question generally to a variety of diagrams, including evolutionary trees. In evolutionary trees, the causal relationship is descent: the literally physical passing on of genetic and other materials to the next generation during reproduction. When two organisms are linked in an evolutionary tree, it means they share a common ancestor more recently than organisms not linked at that level in the tree. Thus, in the tree from Beautiful Evidence, Aotus and Saimiri share a more recent common ancestor with each other than they do with Dolichocebus; also Aotus shares a more recent common ancestor with Teilhardina than with Lemur. (Occasionally, tree diagrams of organisms appear in biology that are not intended to represent common ancestry, but some other property, such as physical similarity. Thus a tree diagram intended to summarize body shape might link sharks, whales and icthyosaurs, although they are, respectively, fish, mammals, and reptiles, and not especially close in terms of common ancestry. Such diagrams are usually called phenetic trees.) It is important to remember two things about evolutionary trees: they are generalized, and they are estimates.
By generalized, I mean that evolutionary history (also called phylogeny) consists of many things, and not all of them can be represented in a single diagram. Generalization is a concept from cartography, and was introduced into biology by R.J. O’Hara (his original paper is available at http://rjohara.net/cv/1993SB.html ). Among the aspects of evolutionary history that we may wish to represent in a diagram are the relative sequence of splitting of lineages (cladogenesis), the amount and kinds of morphological or genetic changes occurring within lineages (anagenesis), the absolute (as opposed to relative) timing of events (absolute chronology), and the geographic distribution of the organisms. It is usually difficult to represent all of these things in a single diagram. The diagram from Beautiful Evidence presents primarily two things: relative sequence of splitting, and the amount of morphological change in the lineages. The sequence of splitting is portrayed by the splitting of the branches in the tree, while the amount of change is represented by the branch lengths. (The diagram is thus not, strictly speaking, a cladogram, which portrays only branching sequence, but a phylogenetic tree. Usage of these terms is not entirely standardized among biologists.) The tree also shows some behvioral information (nocturnal vs. diurnal), but it’s important to note that this distribution is inferred for the extinct forms. There is information about absolute chronology and geography in the text (Teilhardina is from China, and it’s 55 million years old). The names of the organisms encode an awful lot of information, too, for those who know them: for example, I know that Lemur catta is an extant species from Madagascar. For a paleoprimatologist, each name would convey the same sort of information. But such information is not evident in the diagram to the nonspecialist (although the names do allow the nonspecialist to look the information up).
The fact that a phylogenetic tree must be generalized (i.e. some information must be left out) means that an author must choose what to include, and how to represent it. The intended audience, and convention, thus have a significant influence on the representation. One convention that is so universal as to require no special mention is that descent is represented by single lines, rather than by a network of lines representing mating and reproduction (as they would in a pedigree). All primates we know of are sexual, and the inference that the extinct forms were sexual is well supported. The network of mating is generalized to a single line, much as a highway on a map may appear as a single line without showing all the lanes and ramps (a single line in a phylogenetic tree would be literally true only for a wholly asexual lineage). This generalization is so obvious that even readers unfamiliar with such trees readily grasp it. Other conventions are less universal, but they generally do not impede understanding: for example some phylogenetic trees are drawn with V-shaped nodes between branches (rather than the L-shaped nodes shown in Beautiful Evidence). Representing amount of divergence by branch length is a common convention, but the amount must be scaled (as is done in this case), and divergence of what (morphology? proteins? DNA?) indicated; the latter is usually given in a caption, table or text, but sometimes in the diagram itself. Nor is branch length the only way of representing divergence of character. Another very common way to represent it is to put tick marks (sometimes labeled by the feature’s name)on each branch representing the features that have changed in that branch. The less specialist the audience, the more explicitly the conventions should be spelled (or drawn) out. When the organisms are unfamiliar, sometimes a small sketch of the organism accompanies its name in the diagram (impractical in this case, given the large number of organisms). So, phylogenetic trees can be chock full of information, there are many conventions and variations on how to present the information, the more familiar a reader is with the conventions and subject matter the more information they will get out of the tree, but even the best trees have to leave information out (for reasons of space, clarity, complexity, and the fact there are lots of things we don’t know).
In saying phylogenetic trees are estimates, I mean estimates in the formal statistical sense of estimation. The phylogenetic tree shown is the authors’ best estimate of the phylogeny of the animals they studied, based on the characteristics they studied. As anyone familiar with statistics will know, an estimate is obtained by some particular method of inference, and further, there is an uncertainty associated with the estimate: while our estimate may be more or less true, that also means it’s less or more wrong, and we want some way of assessing the strength of our support for an estimate. For example, if we are interested in the central tendency of some numerical quantity, we might use the mean, the median, or the mode: each can be thought of as an estimator of central tendency. For a property like height, which tends to be normally distributed, we would probably regard any of the three as adequate in a very large sample, but for various reasons would prefer the mean in smaller samples. But for a variable like income, which tends not to be normally distributed, we might regard the median as a more useful statistic. For a variable like height, where we regard the mean as preferable, simply calculating the mean is not where we stop. Suppose the mean in a sample is 170 cm. Is this the truth? We might regard it as true for our sample (although, on a slightly philosophical note, I regard even direct observation and measurement subject to residual uncertainty and reexamination: there are some empirical things we can be awfully darn sure of, but nothing that we can be 100% sure of). But if we are interested in height in general, than 170 is an estimate that may be good or bad for all sorts of reasons. 165 cm, or 175 cm, might also be quite reasonable estimates of height in general, although perhaps not as good as 170. We can express our uncertainty in various ways– likelihood, confidence, probability– and many ways would make some use of the standard deviation.
Phylogeny, as something to be estimated, is no different than height or income. There are a variety of methods of phylogenetic inference, like means and medians for height or income. The paper cited by ET uses the method of maximum parsimony. There are others: compatibility, maximum likelihood, Bayesian methods, etc. Particular methods are good for particular types of data. For example, there is a particular kind of genetic sequence used in some phylogenetic studies called a restriction site. We know from genetics that it is hard for an organism to get a particular restriction site sequence (because it requires a whole bunch of genetic changes), but easy to lose it once you’ve got it (because any of several genetic changes will eliminate it). Thus there is a parsimony method that is used for restriction site data that allows loss to occur easily, but gain of a site to be difficult; it differs from standard parsimony in that it attaches different weights to the two kinds of changes that can occur, based on what we know about how genetic sequences mutate. Just as with measures of central tendency, some methods of phylogenetic inference are more or less appropriate depending on the kind of data we have.
The method of maximum parsimony has been much discussed, and is generally thought to be appropriate for the kind of data used in the paper cited by ET. (There is a school of thought that holds that maximum parsimony is always the best method of phylogenetic inference; I and many others disagree, but there is a broad consensus that maximum parsimony is a good method in a large number of situations.) Thus for readers of the paper who are biologists, there is no surprise, and little controversy in the choice of method, in the same way that it would be no surprise, and of little controversy, for an economist to read a paper that used inflation adjusted dollars. The key assumption in the maximum parsimony method is that inheritance is more common than modification (i.e. change in an organism is rare relative to remaining the same). This assumption is certainly supported by what we know about the genetics of mutations and populations, but we can conceive of situations where this is not true, and in such situations maximum parsimony does not work (in the sense that the estimates it provides would not tend toward the actual phylogenetic tree). Indeed, we can conceive of physical processes where it is impossible to estimate the history– consider a marble resting at the center of a soup bowl that slid down from the edge of the bowl: the marble’s current position provides no information on its starting point. But biological reproduction, like xeroxing and medieval manuscript copying, is a proces that preserves a great deal of evidence about its history.
Other methods of phylogenetic inference posit somewhat different processes; the variant parsimony method I mentioned for restriction site data supposes gains of sites much rarer than losses, for example. Debate about the appropriateness of a method usually centers on the validity of its assumptions.
There are two approaches used to express the uncertainty of phylogenetic inferences. One is to try a number of different methods of inference, each with varying assumptions (or else to vary aspects within a method, such as varying the weights given to gains and losses). If the results are the same or similar under a variety of methods, then the results are usually considered more robust, in that they are insensitive to some particular assumption of a method. When the results differ widely, and especially if no method of inference can be argued to be superior on other grounds, multiple trees may be presented. Areas of agreement between trees produced by different methods may be expressed by showing a consensus tree. A consensus tree is a tree that includes only those linkages among taxa that are present in all (or most, or some stated criterion) of the trees produced by the varying methods. Far from being some sort of anti-empirical hocus pocus, the use of a consensus tree expresses the uncertainty about the results: well supported relationships are shown, but poorly supported relationships are collapsed into unresolved branching points, meaning that the data and methods of analysis were unable to say what the relationships are at that point.
The second approach is to explore the uncertainty surrounding an estimated tree obtained by a particular method. The authors of this paper found 33 equally parsimonious trees. That is, based on their data they could not distinguish between 33 trees, each of which was equally well supported. What they did was not to arbitrarily choose among them, but rather present a consensus tree, which has the exact same properties as does a consensus tree based on a consensus among methods (in the paragraph above). Thus far from hiding the alternative trees, they are actually all right in the diagram! The individual alternative trees, which can be obtained by resolving the polytomous branch points into series of dichotomous branch points (you are also likely to obtain some slightly less than maximally parsimonious alternatives among the many possible resolutions), are not shown because it would require much comparison among them to detect their points of disagreement, while the consensus tree readily identifies for the reader the parts of the tree that are most uncertain. Other ways of assessing uncertainty are also widely used: bootstrapping, jackknifing, and parametric approaches as well. When bootstrapping is done, it is commonplace to have each node in the tree accompanied by a a number in the diagram indicating the percentage of bootstrap replicates in which a grouping occurred. One feels more confident about a branch with, say, 99% support, than one with 50% support.
As regards ET’s specific “built-in assumptions”, 1) there is nothing in this tree which assumes (or supports) a single origin of life on earth. There are a variety of arguments in favor of that proposition, and in certain phylogenetic studies of bacteria it may be assumed, but this study of primates only deals with common ancestry in this group of mammals (plus a few close relatives at the base of the tree); 2) the tree in fact is not dichotomous– the polytomies reveal the uncertainties associated with the best estimate of the phylogeny. Multiple branching is in fact perfectly possible with the method used by the authors, but in a large data set, even true multiple branch points tend to be resolved by random differences; there are ways of checking whether a dichotomous branch point is real or a statistical fluke; 3) yes, this tree does show descent with modification (although as noted before, there are certain special trees that do not reflect descent with modification, and thus don’t make this assumption); 4) the use of parsimony, while not present in the diagram, is stated in the figure legend (which also explains the meaning of daggers and asterisks), and also in the methods section of the paper, where the exact computer program used is also specified. The various statistics given in the figure legend give a further indication of the uncertainty of the results. As regards the breaking of ties, unless the authors have done something not evident from their paper, I’m not sure what averaging method ET refers to. Consenus trees are not a tie-breaking method, but a way of showing what is well supported, and where the uncertainty lies.
So, is this an evidence-thin diagram selected by some arbitrary method? The answer is no. The data matrix contains 303 organismal characteristics assessed for 52 taxa, or over 1500 entries. Each characteristic is some observed or measured feature of the organisms. The method of inference is a standard one, whose properties have been much studied; its most ardent users prefer it because it, in may ways, makes the most minimal assumptions about desent and modification. When the tree inference method cannot decide among competing trees (as in this case with 33 equally parsimonious trees), the consensus tree method allows those parts of the estimate that are well supported to be shown, while pointing out the areas of uncertainty. The particular consensus method chosen by the authors, called strict consensus, is the most rigorous of consensus methods, in that if even one of the 33 trees disagrees with a linkage found in the other 32, the tree is collapsed at that point into a polytomy, revealing the uncertainty. Far from alternative trees being absent, the authors have given the strongest influence to each alternative, and pointed out where the uncertainties are (that’s what the polytomies are). The strengths of this study, and the resulting tree, are the large amount of data, and the fact that the strict consensus shows so little disagreement among the 33 trees.
What valid criticsms might be made of this paper and tree? First, the data are not given; one must go to Nature’s website, to find the data in a file there. I particularly dislike this practice, because the data will therefore eventually probably become lost or unavailable, and it is in scrutinizing the anatomical details in their data where most of the biological discussion lies. Nature, however, would never publish a paper that had so much data, for reasons of length. (As an aside, both Science and Nature are like this: they won’t publish data-rich papers, at least not if the data is in the paper. Some years ago Jared Diamond published a 10 page paper in Science on the ecology and distribution of New Guinea birds based on many years of field work, but the data were published in a 400 page book.) Second, there are other ways of exploring uncertainty in the estimated phylogenetic tree that could have been used: bootstrapping, for example. Another way, less frequently used, would be to use not only the most parsimonious trees, but also slightly less parsimonious trees in arriving at the consensus. Third, and trivially, the dagger symbol is a conventional indication that an organism is extinct, but it is used here in a somewhat different way that seems odd when glancing at the figure, but the usage is explained in the figure legend.
A more general criticism might be that, despite using a well known method, the authors might not fully understand the assumptions and limitations of the method. Such has certainly happened in the past. But there is nothing to indicate that is the case here. Someone may use a t-test badly, but that doesn’t mean that every author who uses a t-test does, nor that a full discussion of the assumptions and limitations of parametric statistics should accompany each use.
I have taken up ET’s challenge to be as demanding of the criticism of the diagram as of the diagram itself. I do not believe the issues raised in the criticism are serious. This does not mean that ET is a “whiny critic”. I have been impressed, and even moved, too often by his careful analyses to dismiss his critique in this way. The study of methods of inferring phylogeny is a very important part of biology, to which much effort has been devoted, and much work is still being done. The problem of representing phylogeny in diagrams goes back over 150 years, and has a rich and diverse history. There is much that can be done, and application of ET’s principles will, I am sure, have a salutary effect. It is difficult, however, to come into a field with such a long history of methodological development and representation, and immediately see what the issues are. The critique fails to see that a consensus tree is precisely the presentation of alternative trees that ET seems to desire. Even a Berkeley website may not provide sufficient background.
I’ve attempted to make my points here using simple statistical analogies because I think most readers of the forum are statistically literate, and can appreciate that there is a great deal of statistical theory and debate, underlying even simple things like means, medians, t-tests and inflation adjustment. Phylogenetics has a similarly rich underlying basis. The book Molecular Evolution: A Phylogenetic Approach, by Rod Page and Edward Holmes (1998, Blackwell; http://www.blackwellpublishing.com/book.asp?ref=0865428891&site=1 ) has a readable introduction to many issues of inference and representation. (Page also has a free program called Tree View at his website.) For the most comprehensive discussion of the assumptions and justifications of methods of phylogenetic inference, see Inferring Phylogenies, by Joe Felsenstein (2004, Sinauer; http://www.sinauer.com/detail.php?id=1775 ). The latter is based on a very deep understanding of the methods, and the need for drawing out the implications and assumptions of each method (the latter stems from Felsenstein’s statistical approach to phylogenetic inference, a view I share, but which is not universal among biologists).
This is an interesting contribution by Kindly Contributor Gregory Mayer. Thank you.
I have some questions, some of which are surely foolish or impossible:
(1) Where in the tree pictured can the viewer see estimates of uncertainty?
(2) What counts as a bifurcation and what counts as a multiple simultaneous division? How many of each occur in this tree?
(3) Are there trees for these data calculated prior to the recent discovery? How do the prior trees differ from this one? How should they differ at all, with the exception of placement of the new discovery?
(4) Is there any substantive defense of equal weights for all characteristics?
(5) In thinking about the size of data matrix (small compared to similar data in, say, survey research or psychological testing), are the entries assumed to be independent?
(6) Suppose all 31 maximally parsimonious trees were printed on transparent paper and piled on top of each other. How blurry would the cumulative image be?
My purpose in this BE chapter on causal arrows and linking lines is to suggest methods for examining and cross-examining trees from any field. That is, how well does a tree meet universal explanatory standards? (This says a lot about how trees should be designed.) An answer to that question will come from the match of universal explanatory standards with the performance of the tree, rather than with reference exclusively to current practices within the field. Some disciplines may have a bias toward toward over-fitting empirical data to produce brittle models (as in econometrics or factor analysis), toward adopting new modelling assumptions when new evidence is scarce, and toward self-congratulation.
The issue is the external validity of the tree, not just the internal validity within the discipline. That is how I would evaluate a postmodern lit-crit analysis of something, rather than asking if the postmodern analysis was consistent with the current intellectual practices among postmodernites.
Beginning as a good example because of several good design features, the tree wilted somewhat as I read the fine print about the underlying methods. Maybe the tree will perk up again after this tutorial.
(1) Where in the tree pictured can the viewer see estimates of uncertainty?
This particular tree does not seem to have any. In other trees, as
Gregory Mayer noted, you will see numbers next to the branches / nodes of the tree to indicate degree of uncertainty.
Some researchers, however, contend that trees should contain only bifurcations (on the assumption that evolution proceeds only by bifurcation); such individuals would argue that any simultaneous divisions (below) would represent an area of insufficient data rather than a real biological split of one species into three.
(2) What counts as a bifurcation and what counts as a multiple simultaneous division? How many of each occur in this tree?
I count five non-birfurcations (polyfurcations?). One, at the base of the tree, contains Scandentia, Purgatorius and “everything else.” Another, in the middle of the tree contains Nannopithex, Trogolemur, Psuedoloris and one other branch that then bifucates. Bifurcations have two line segments leading to the right from each horizontal line in this style of tree; simultaneous divisions have multiple line segments leading to the right from a horizontal line.
(3) Are there trees for these data calculated prior to the recent discovery? How do the prior trees differ from this one? How should they differ at all, with the exception of placement of the new discovery?
About this particular case, I cannot say, but there probably would have been other phylogenies, although perhaps not as detailed. The last question is the key one, and yes, adding new species can change the entire topology of the tree.
(4) Is there any substantive defense of equal weights for all characteristics?
There is a large techical literature addressing this question. The argument for it is that by weighting characters, you are incorporating assumptions that some features are more evolutionarily plastic than others. That should be an hypothesis to be tested by the tree, not an assumption used to build the tree.
(5) In thinking about the size of data matrix (small compared to similar data in, say, survey research or psychological testing), are the entries assumed to be independent?
I think so, but I’m not sure. When people are comparing different species for purposes other than building a tree, however, there are corrections to account for the fact that related species are not independent samples, based on their shared evolutionary history.
(6) Suppose all 31 maximally parsimonious trees were printed on transparent paper and piled on top of each other. How blurry would the cumulative image be?
I suspect that you’d only see the blurring at the points where there are not bifurcations.
The issue is the external validity of the tree, not just the internal validity within the discipline.
For that reason, I would like to emphasize that the tree represents an hypothesis. (I think Gregory Mayer said much the same thing except using the term “estimate” instead.) A tree is useful if it has predictive power. For example, there are three species of Teilhardina in the tree pictured. Two belong to a single group, while a third (T. americana) belongs to another cluster. This suggests that T. americana might actually be better categorized as another genus, and that T. americana is more likely to share features with, say, Necrolemur than Tarsius. If the tree fails to predict the distribution of characters not in the tree, it’s time to re-examine the tree.
One other point to make about the tree shown is that it treats extinct and extant species the same way. Both are shown as terminal branches on the tree. There are no species named back in the nodes. But evolution means that one species can literally become another. There isn’t, as far as I know, a rigourous way to construct a tree where one species can be hypothesized to be an ancestor of another. The analysis effectively treats them all as contemporaries.
I am happy to help with ET’s tutorial in phylogenetics. It’s useful to me (since I have to explain these things in my teaching), and I hope it’s useful to others as well. Before getting to ET’s 6 new questions, I want to remark on his broader project, which is to consider tree diagrams in general, and how we evaluate them. I find this a very fascinating area, and participated in some very interesting discussions on the matter a few years ago.
My remarks on the broader project are confined to a subset of tree diagrams– trees which depict descent with modification. Such trees are not limited to biology, but are an important component of at least two other fields: stemmatics and historical linguistics. Stemmatics is the study of the history of the copying of manuscripts (hence my allusion to medieval manuscript copying in my first post). Stemmaticists carefully study texts, and attempt to determine which copies were made from which other copies. The copying is usually traced by the introduction of small copyist’s errors, which are then perpetuated in any copies descended from the one in which the error arose. Tracing these small errors allows one to trace the history of the manuscripts. For example, if a copyist changed “Beowulf” to “Biowulf”, then subsequent copies made from this one and its copies would also have “Biowulf”, and all these could be inferred to have descended from a common copy. (Beowulf is known from only one copy, so the example is entirely hypothetical.) It’s possible, of course, for a copyist to realize an error has been made, and thus switch back to “Beowulf”. This complicates things, and that’s what makes stemmatics interesting– I can only scratch the surface of the process of copying, and how stemmaticists infer the history. Stemmaticists make tree diagrams of the relationships among copies that look very much like the kind used by biologists.
Historical linguistics is probably more familiar: we all know how the Latin of the late Roman empire changed and diverged over time to form the current languages of Italian, French, Spanish, etc. Historical linguists trace the changes in languages, sometimes by looking at the extant languages, but also by looking at old texts (which are like linguistic fossils), in which case they may also be aided by stemmaticists. Historical linguists also make tree diagrams much like biologists do.
There have recently been some attempts for cross communication and cross fertilization of these fields. Much of the effort has gone into exploring the analaogies and disanalogies of process among the three fields. The copyists’ errors in stemmatics are like genetic mutation in biology; the correction of errors corresponds to back mutation. Word borrowing is like hybridization in biology. A difference is that hybridization is much more rampant in languages (think of English after the Norman invasion) than among biological species. Linguistics tends to have more process rules (or assumptions), such as laws of sound change, than the other two.
I’m not sure what other kinds of trees ET had in mind, but these kinds of trees– called “Trees of History” by Bob O’Hara– have many points of analogy, including methods of inference, and would be interesting to compare. O’Hara hosted an email discussion list from 1993-1997 on the historical sciences, and it is archived at his website ( http://rjohara.net/darwin/ ); much of the discussion is on the different “tree making” disciplines. Also at his site is his paper where he coined the term “trees of history” ( http://rjohara.net/cv/1996Milan.html ; see the pdf for the figures). (Suggestion for ET: O’Hara is at Middlebury in Vermont– you should go up and talk to him.)
Now for the 6 questions.
1) Where are the estimates of uncertainty? They’re right in the picture– the (by my count) 5 places where a polytomy (a split into more than two branches) occurs. For example, near the middle of the tree, Microchoerus and Necrolemur are each other’s closest relatives, but the tree reveals uncertainty as to who their next closest relative is: it could be Nannopithex, Trogolemur, or Pseudoloris. We’re also uncertain as to how these latter three are related to the first two or to one another. This means that in all 33 trees the first two were each other’s closest relatives, while there was at least one tree that disagreed with the other trees regarding the relationship of these two to the latter three.
The consistency index and retention index (in the figure legend) are two quantitative measures of how well the characters (i.e. the data) fit the tree: the larger the numbers, the better the fit. The consistency index is the minimum number of changes in characters (in this tree 765) divided by the actual number of changes in the tree (in this case 2076). If the consistency index were 1.00, all the characters would perfectly fit the tree. When it’s less than 1.00, there is what is called homoplasy in the data: the need to invoke convergence in character and reversals in character in order to fit characters to the tree. When the consistency index is 1.00, there is no need to invoke parsimony, because all the data are in agreement; parsimony is a method of resolving conflict between characters as to which tree is correct. The consistency index is known to scale downward as the number of taxa and characters increase, so it is difficult to use for comparison among data sets of different sizes. The retention index is an attempt to make a more data-set size independent measure of homoplasy. It’s formula is a bit more complex, but suffice it to say it should be gretaer than the consistency index, and it is. There is no theory of what the distribution of these indices should be, and thus no parametric-type statistical tests are possible. Permutation tests are possible, and they have shown that phylogenetic structure is present (as opposed to a random distribution of characters) in data with consistency indices below .3. Thus this study’s indices would be judged to be alright. I know ET doesn’t want to know just what a biologist would think, but what I’ve tried to show is that “alright” is not what a biologist would “just think”, but rather it would be a judgement based on serious attempts to find ways of measuring the agreement between data and estimate, and the use of statistical tests to evaluate the measure.
Thus how confident we should be in this tree is expressed in two ways: in the tree itself, the exact points of uncertainty are highlighted by the polytomies; and in the indices, we have an overall measure of the fit od the data.
2) What is a bifurcation? What is a polytomy? A bifurcation is when a vertical line gives rise (to the right; all orientations are relative to this diagram, which of course would differ for other ways of laying the tree out on a page) to two (and only two) horizontal lines. A horizontal line can be of 0 length (note Teilhardina belgica just above T. asiatica). A polytomy or simultaneous multiple branching occurs when a vertical line gives rise to 3 or more horizontal lines. There are about 5 polytomies, and lots of bifurcations in the tree. Polytomies can signify one of two things: either uncertainty about how to resolve them into a bifurcating tree (these are called soft polytomies), or that there really was a simultaneous split of one lineage into three or more lineages (hard polytomies). Hard (or real polytomies are entirely possible. For example, I study lizards which lived on a big island which broke up, essentially simultaneously, into several smaller islands as the ice age glaciers melted and flooded the lowlands (we call these smaller islands the Virgin Islands today). There is thus a hard polytomy among the lizard populations on these islands. The method of parsimony is entirely compatible with hard polytomies, although, as I mentioned previously, one rarely finds them in trees produced by the method, since, in a large data set, random similarities will often separate the several branches (and it is possible to test for this). In the primates we are dealing with here, the polytomies are soft, produced by the consensus method, and reflect uncertainty.
I’ve run out of time (off to a meeting), and will address the other questions later. I do want to point out before I go that ET changed the BE text above after my original post. In particular, I want to point out to readers first seeing this that ET used the phrase “whiny critic” in his original text, and I was merely using his phrase: I intended no insult or disrespect by its use.
A continuation of my response to ET’s questions.
3) Are there prior trees for these data? Yes. Most of the data, in fact, comes from an earlier study (#21 in the list of references in the original paper). This is a much longer paper (nearly 90 pp.) in the Journal of Human Evolution. I do not have immediate access to the paper, but from the online abstract I can surmise that this paper includes a much more careful anatomical consideration of the data matrix, and includes a lot more variations in how the data were analyzed, and almost certainly has a number of trees. The abstract refers repeatedly to relationships that are well supported, and others that are uncertain or poorly supported. Since I haven’t seen the whole paper, I don’t know where the previous trees and this one might disagree. I’m sure the Yale libray must get the journal. People really interested in primate phylogeny will want this paper, because it will deal with many issues that are necessarily left out of the Nature paper, due to space limitations. As I mentioned earlier, most of the criticism and discussion of a paper of this kind revolve around the character analysis that went into the data matrix.
Papers in regular systematics journals tend to have more trees. For example, the first article in the October 2003 issue of Systematic Biology that happened to be on my desk this morning has 16 trees in a 22 page paper (the paper looked at about 3000 molecular characters in 24 small carnivores). (Yale gets Systematic Biology.)
As regards the question of what effect the new discovery might have on the previous tree, the answer is maybe none (other than its placement on the tree), or maybe a lot (although with only one taxon and 12 characters added, I would be surprized if there was a large effect). One of the methods of assessing uncertainty in a tree estimate is jackknifing on taxa, in which taxa are removed from the analysis one (or sometimes two or more) at a time, and the result of their absence noted. The more varying the trees produced by jackknifing, the less certain we are in the tree. The prior tree is like a jackknife tree of the new tree, with the new species being the deleted taxon. There was some debate a few years ago about whether the inclusion of fossil taxa could alter the analysis, and it’s pretty clear now the answer is that they can.
4) Is there a substantive argument for equal weighting? In the context of phylogenetic inference, I would say there is not a substantive argument for equal weight. The argument for it is methodological: how would we know what weights to use? Or, in slightly different form, to weight the characters would be including unwarranted assumptions in the analysis. There are a number of ways of weighting available. No way of applying them to morphological characters seems entirely successful. However, for molecular data, there are a number of widely used weighting schemes, mostly based on the idea that slowly changing genetic sequences are more informative (hence have higher weight) than rapidly evolving ones (which hence have lower weight). The principle is the same as in the restriction site analysis I mentioned in my first post. A low probability change (i.e. difficult to happen or occurring rarely) is less likely to have occurred twice, and thus organisms sharing it are more likely to have inherited it from a common ancestor than to have evolved it independently of one another.
One of the problems of molecular data is that the range of each character is limited: there are only four nucleotides in DNA. This leads to the phenomenon known as saturation: if a sequence has changed enough, there will be an asymptotic similarity of 25% of all sequences. When this happens, the history is unrecoverable (its like a marble resting at the bottom of a bowl– you can’t tell which side it rolled down). So, for DNA data, it is especially important to downweight sites that are evolving too rapidly, since there is no phylogenetic “signal” among the noise. One reason that morphological characters tend to seem reasonable under equal weighting is that morphology is much less constrained in its range than DNA (compare a lamprey to an elephant among vertebrates), and so tends not to “saturate”.
5) Is the data set small? Well, there are 15,000 cells in the data matrix (I was off by a 0 when I wrote 1500 previously). This seems pretty large to me. I’m not familiar with survey research (other than having been polled over the phone occasionally!), so I don’t know how it compares. The Challenger graph on p. 45 of Visual Explanations only had 23 points, and I was convinced.
Are the data independent? This is a very interesting question. In order to have the greatest confidence in our phylogenetic reconstructions, what we want is for each character to be evolving independently of every other character. That way, each character provides independent evidence. For instance, suppose the length of the right leg was used as a character. We then would probably not want to use the length of the left leg as a character, because, given the general bilateral symmetry of vertebrates, the two legs do not evolve independently of one another. The length of the bill (let’s say we’re thinking of a bird), however, is probably independent in a genetic-developmental sense (except as both bill and leg are affected by overall size). But because both bill and leg occur together in the same organism, they will come to be correlated in their occurrence with one another. It is precisely this latter nonindependence, which is caused by descent with modification, that we are trying to detect. It is analogous to when we are examining a possible correlation between two variables. We want our data points to be independent, but when the analysis is over and we find a significant correlation, it turns out that the variables are not independent. So, yes if you know the value of one variable, you can often know the value of others. If you tell me something has feathers, I can tell you a lot about its feet and skull (to use another bird example).
6) What would happen if you overlaid the 33 most parsimonious trees? There would be very little blurring. The only blurring would occur in the immediate vicinity of the polytomies. This, as I’ve mentioned before, is a strength of this result: the lack of consensus was localized to a few small areas of the tree.
If, as an earlier comment seems to indicate, ET suspects phylogenetic inference is a species of postmodernism (i.e. a self-referential system disconnected from the empirical world), then I have utterly failed in my attempts to explain some of the issues involved. I would prefer to think that is due to my explanatory inadequacy. While ordering from Amazon, in addition to the two books I mentioned previously, I would suggest Reconstructing the Past, by Elliott Sober (1988, MIT Press http://mitpress.mit.edu/catalog/item/default.asp?sid=D54454DD-6B4E-411D-B0D6-4F56CB76C0BF&ttype=2&tid=6625 ), a philosopher who is, in general, much better at explaining things than I am. I would be glad, however, to answer further questions.
A point I left out in response to question 1) on uncertainty: There is another indication of uncertainty in the branch lengths. One way of stating how well supported a particular relationship on the tree is by stating how many characters support it, that is how many evolutionary changes are in the branch that unites them. Because the horizontal branch lengths are scaled to the number of characters, then shorter branches are less well supported.
Greg Mayer called my attention to this discussion. ET had raised many of the relevant questions, and Greg and Zen Faulkes have pointed out the ways in which the literature in recent years has tried to answer them. I would add (self-servingly) that my recent book Inferring Phylogenies (Sinauer Associates, 2004) will serve as a more detailed reference for the field. In particular, in Chapter 34 I agonize over how to draw phylogenies (evolutionary trees) best. They are inherently high-dimensional, and summarizing their statistical uncertainty is even more complicated. If no one two-dimensional diagram suffices, that should not be surprising. Any flat representation inevitably conveys to the viewer some misleading information. For example, the order of tips across the rightmost (or topmost) part of the tree can be changed by flipping the order of branches left-right at the forks. The results are all supposed to represent the same tree, but may seem to imply different closenesses of the species to the unwary. Likewise, choosing to have the fork be square versus V-shaped implies to the viewer sudden or gradual divergence.
Gregory Mayer clearly has a deep understanding of phylogenetics and I defer to his excellent analysis. I have two additional points–one on the choice between many competing trees, and one on external/internal validity.
I would like to reiterate that most phylogenetic trees are hypotheses based on the best available evidence. The question that we must ask as viewers of the tree: is the evidence used to its best potential and not beyond? This tree is remarkable in the resolution of its lineages. Oftentimes there are many more unresolved lineages like the one that includes Nannopithex, Trogolemur, Pseudolaris, and the Microchoerus/Necrolemur lineage. This tree appears so well resolved because someone spent a tremendous amount of time at a microscope measuring 303 characters on all 52 species. The biggest assumption behind the accuracy of the tree is that these 303 characters are inherited, do not evolve too quickly, and are independent of one another. The criticism that seems most valid to me is that some of those 303 characters may covary with each other (a whopping 194 were dental characters) and thus the analysis might include pseudoreplication (see Mayer’s example of right and left leg). Thus, it is possible that the authors have overtaxed the evidence. The vagaries of fossil evidence may not allow such a fine scale resolution of descent, especially when relying on a chunk of fossilized skull or some other incomplete fossil instead of the whole organism. Of course, I have not picked apart every character that went into the analysis, which would be the proper way to judge these findings. Maybe I owe my dentist a renewed respect for mastering all 194 distinct features of my primate dentition.
Here’s a question that analyses like this one bring up for me. Are there only 33 trees we should consider out of the 2.7 x 10^78 possible ones with 52 taxa? To understand what the authors mean when they say that they generated 33 equally parsimonious trees, it helps to know how the trees are quantitatively generated (a common undergraduate exercise: here’s an example) and then to see one of the computer programs (e.g., PAUP) that generates these trees in action. An interesting point that Mayer mentioned is that although there are 33 trees that are equally parsimonious, there are an untold number of other trees that are almost as parsimonious. The inclusion of these in the strict consensus tree would increase uncertainty and perhaps rightly so. Maybe systematists should think of a way to include this type of uncertainty, like a strict consensus of the top 5% of most parsimonious trees for example. It seems to me that from an evidence presentation standpoint, the number 33 is not very informative and is a little arbitrary. Furthermore it is confusing, even misleading, to anyone who has not seen how PAUP or other phylogeny inference software works, which brings me to the external/internal validity debate!
Perhaps this is another thread for this board, but it seems that especially in high profile publications there can be a tradeoff between accessibility, brevity, and rigor (a.k.a. external validity) in data presentation. Like time, money, and quality, you pick two of three. For example, although the general scientific audience for which Nature publishes may not know what a consistency index or retention index is (inaccessible), Ni et al. must include them for rigor, but cannot explain them because that would sacrifice brevity. To cover its bases, the news section of Nature emphasizes accessibility at the expense of rigor. It does this by by eliminating discipline-specific codes for quality ensuring procedures like “retention index.” (Also, same goes for other popularizations such as the New York Times tree we discussed in a previous thread.) I’m thinking that reduction in accessibility doesn’t necessarily decrease external validity–it’s just annoying to outside readers–but perhaps that’s debatable.
Systematics is an exciting field with interesting visual representation challenges. I am excited to see E.T. tackling these tough issues!
I have three questions, which may be on or off the wall.
(1) TIME So the time of a split is strictly a locally ordinal variable in the Nature-article tree? That is, as we move left to right in looking at the tree, we have an order of animal appearances but only an order. And is that ordering local to a particular branch–that is we can’t compare the time (years, not number of bifurcations) of a noun’s appearance on one branch with the time of another noun’s appearance on another branch? To put it another way, nouns that are vertically aligned may differ greatly in when they actually appeared on earth? If this is true, that is giving up a lot visually, a big variable, the estimated time of the appearance on the noun on earth. After all, one of the Nature article’s major points is that the skull has an estimated age of 54.97 Myr ago (there are those pesky 4 significant digits again) or in 54,968,000 BCE. It would be helpful to see the other nouns dated on the tree (presented in the data base?). Also a time scaling somewhere on the graphic would demonstrate how amazing the new discovery is, by showing a comparison of the newly found skull with all the past finds. How about an Myr date by each noun?
(2) HORIZONTAL LINE LENGTH Since the length of a link is the number of features (same as characters?) tied to the noun at the right end of the link, how can that link-length be called a “measure of uncertainty”? It is simply the the number of features; how can it be turned into a statistical virtue, especially since the features, at least at times, are not independent of one another. So the length of the line reports on the completeness of the fossil (perhaps a measure of uncertainty) and on the industriousness of fossil measurement. But all those dental measurements may inflate the N, and therefore the length of the line. How about making line length proportional to log N? Or use a statistical technique to dilute the intercorrelation and deflate the N?
(3)VERTICAL LINE LENGTH Any thing going on here?
.
Cuthbert Daniel, the great contributor to industrial and process manangement statistics, once described “bang-bang duplicates,” in which an observer looks at a dial, writes down a number, and, a few seconds later, looks at the dial again, and writes down a second number. The N here is not 2, it is probably more like 1.02. That second observation has probably brought in very little new information. Is that what you man by “pseudoreplication”? To put it another way: no field underestimates its sample size, and the assumption of independence always exaggerates effective sample size (although in econometrics there is a lot of work on reducing spuriously large Ns in time-series by figuring out autocorrelation in a sequence of observations). By the way, the independence-of-observations assumption shows up all over the place and leads to underestimates of error (e.g., in survey research, with inevitable clustered samples, the N for computing sampling variation is taken as the full sample size in most practical applications, as in the +/- 3% statements in newspaper surveys).
On equal weighting. John Tukey once remarked that all weightings are arbitrary, including equal weightings, and that we should be wisely arbitrary. Bob Abelson and John Tukey wrote a couple of papers years ago on generating weights for categoric data.
Dear Contributors, I appreciate your most helpful comments so far and your reading assignments. My visit to cladograms is to show BE readers how to read and reason about trees, any trees–and not to criticize or dare to contribute to an obviously smart and thoughtful field. Thank you for helping an outsider poking around like this. ET
It would be a reaonable hypothesis that the tree represents an order of appearance, but that is not necessarily the case. As I mentioned in on of my earlier posts, notice that the extinct species are intermingled with living species on the tree. Incorporating fossil data really poses a problem with phylogenetic reconstruction.
Imagine if you tried to build a family tree based only on physical resemblance. It would be a reasonable to think that the more closely related members of the family look alike. Now imagine that you tried to incorporate several generations into this tree — but you didn’t use that generational information, you only used physical resemblance. If you happen to bear a strong resemblance to your grandfather, then comparing physical resemblance alone might suggest you and your grandfather are more closely related (i.e., adjacent branches on a tree) than you are to your father.
That is kind of the situation phylogenetic reconstruction is in. Temporal information is not included in the data for the analysis, so the best you can get is an hypothesis about timing.
Yes, vertically aligned nouns could have appeared at different times in history. What is being shown is the number of character changes in this particular figure, and those morphological changes may not (or may!) occur at uniform rates. Some speciation events are very rapid (African rift lake cichlid fish are a classic example) whereas other groups show relatively little morphological change over long periods of time.
A lot of that information might not be known; there might be no fossil record for the living species to include, and you’d have to make estimates based on phylogenetic trees.
A standard way to do incorporate time is to redraw the tree with time as the X axis. Frequently, solid lines are shown to indicate presence established by fossils, and dashed lines indicate an absence of a fossil record. The original and the new could be shown next to each other as a small multiple.
In one sense, however, the nouns are labelled with temporal information. The crosses indicate extinction; asterisks, extant. Admittedly, it indicates termination rather than origin (the latter typically being of more interest in trees), and it provides only two wide categories (extinct and not-yet-extinct), but it does have the advantage of being concise and adding extra information without making the table difficult to read.
Time is indeed not represented quantitatively in the Ni et al. tree. Splits that are vertically lined up may have occurred at different times in Earth’s history. Some trees do attempt to represent time, like the NYT human evolution tree. When we see these, we should be suspicious of every proposed splitting date, because they are hard to determine. Also, in the news and views section of the same issue of Nature, there is a much less detailed tree summarizing the findings of Ni et al. that attempts to date the more interesting splits (tree also comes complete with chartjunk, weird 3D spheres, and exuberant use of gradients). If every character evolved at the same rate, dates would be easy to determine, but that is far from the case. One of Stephen J. Gould’s biggest contributions to biology was to provide strong evidence that organisms often stay the same for long periods of time before changing rapidly and drastically.
I think what G.M. is saying about branch length is that they provide a qualitative measure of uncertainty because the more changes that occur, the more certain we are that a split has occurred. For example, if you are comparing a golden retriever, a greyhound, and a housecat, the branch that leads to housecat would be longest because the housecat will have more character differences; thus we are more sure that it is a separate lineage. This is generally but not always true because a bunch of changes may occur simultaneously (like if an animal evolves the ability to fly, its anatomy could evolve very quickly to favor the best flyers). G.M., please correct me if my interpretation is wrong.
Nothing going on with vertical line length. The taxa names (in this case all Latin binomials) are evenly leaded and the vertical lines are made however long they need to be in order to connect the branches.
Inflation of sample size by using replicates that are not independent leading to overestimates of certainty is indeed what I meant by pseudoreplication. A colorful paper by ecologist Stuart Hurlbert brought the rampant statistical crime of pseudoreplication in experimental analysis to the attention of ecologists. (Pseudoreplication and the design of ecological field experiments. Ecological Monographs 54(2) 1984. pp. 187-211. Can be retrieved at http://www.jstor.org with subscription.) He would call Daniel’s dial and other types of sequential autocorrelation “temporal pseudoreplication.” Hurlbert’s introduction includes the quote, “I don’t know how anyone can advocate an unpopular cause unless one is either irritating or ineffective. -Bertrand Russell” and he lists “nondemonic intrusion” as one of his keywords!
Interestingly, the author of the news and views summary of Ni et al., Robert Martin, dings the paper for yet another type of pseudoreplication. He calls into question the determination of activity patterns (diurnal v. nocturnal). His rationale is that the regression of orbital diameter against skull length that is supposed to predict activity pattern relies on phylogenetic pseudoreplication (also has been called phylogenetic inertia). This occurs when closely related species such as Proteopithecus sylviae and Catopithecus browni are treated as independent data points. Both might have lower orbital to skull ratios (small eyes) but if small eyes developed in one of Proteopithecus’s ancestors and were simply passed down to other descendants which include Catopithecus, there is only one instance of evolution of smaller eyes, not two. The regression should take that into account. Martin also points out that conclusions about T. asiatica’s activity pattern rely on an extrapolation that is pretty far outside existing data and that this might not be valid. I agree that these concerns cast doubt on T asiatica’s status as a “small, diurnal, visually oriented predator.”
All this said, I think that the conclusion that the T. asiatica skull represents a novel paleontological discovery of one of the earliest primates and that this primate lived in China is still valid and pretty neat.
For this display, then, it is not meaningful to talk of an XY overall scaled grid, since the X scale is locally applied and Y is the leading of the type labelling the nouns. This display resembles a sociogram, with distances between nouns measuring social distances. The tree metaphor (or flow-diagram metaphor) is not quite appropriate here.
Perhaps the first piece of advice to the innocent reader of any tree (and for any graphic) is to figure out the XY grid, if any. What exactly is being measured in the display and how is it shown? For example, in the Barr art chart, it is time flowing downwards; and then left to right, art style (more or less geometric). And for this cladistic diagram, the XY question at least points to an understanding about what is going on. For the cladistic diagram, readers (well, a few of the readers of Nature) are working within a disciplinary context (which, from this thread, has some variability) that provides the necessary background to read the diagram. The naive reader may think time is flowing left to right.
Time data, although scarce, still might be shown when available.
I find Martin’s contributions valuable, particularly the graphic. I think some of his questions were rhetorical, but I’ll take a stab at the regardless.
A good reason why indicating extinct species is helpful! In most cases, however, that sort of source material will probably be described in methods or an appendix rather than being shown in the tree itself.
Did you mean “organismal characteristics?” You do see some trees that have key features shown in the branches between the nodes. I use small, labelled boxes in the branches between branch points. Next, as I noted in an earlier response, weightings are typically equal in this field.
It doesn’t take a new species to challenge the tree. Trees are built from a small set of characters, but if it is a “good” tree, it can predict the distribution of other characters in the species examined. For instance, there are three species at the “top” of the tree shown: Aotus, Saimiri, and Callicebus. If you found a particular character in Aotus and Callicebus, the tree would lead you to predict that you would also find it in Saimiri. If you did not find the predicted character, that might give you reason to doubt the tree. You wouldn’t necessarily revise the tree, because there are many evolutionary reasons that might explain the absence of the feature in that species. You’re in a realm where you can rarely apply a simple experiment aimed at disproving the hypothesis presented in the tree, which is obviously very complex.
That can’t be resolved through this type of analysis, for reasons similar I described in an earlier post about the problems with incorporating extinct species in the tree. It is a limitation of the methodology that has yet to be overcome (as far as I know).
That would have to be answered on a case by case basis. That information probably doesn’t have much bearing on the relationships shown in a tree of this sort, however.
I’m delighted to see Prof. Tufte taking an interest in evolutionary trees. I’m biased, but I think they are among the most subtle and complex diagrammatic representations made, and we’ve still got a lot to learn as we develop sophisticated ways to represent and explain them.
I can’t do a better job than my old colleague from grad school, Greg Mayer, in explaining the principles of phylogenetic inference; I don’t have the mathematical brain that Greg has and Joe Felsenstein has (Joe has been one of the leading contributors to this field for the last 20 years). Instead, I’ve tried to carve out a separate niche for myself in analyzing evolutionary trees as representational devices, rather than taking on the problems of inference. I hope ET will remain interested in this topic, and I think he will find this particular class of diagrams to be very provocative of thought.
I’ll only add a few things to what has already been said, and I’ll make bold to refer to some of my own work because I think it really does address a number of the issues that ET has raised in this thread and elsewhere. Here I’ll focus on the intellectual contexts in which evolutionary trees can be examined; I’ll follow up with a post about representational issues.
Evolutionary trees can be considered in three non-overlapping contexts: (1) as branching diagrams, (2) as diagrams of what is called the Natural System, and (3) as genealogical “trees of history.”
(1) It is as simple branching diagrams that evolutionary trees are least interesting and least appropriately considered. By simple branching diagrams I mean “logical trees,” “decision trees,” “classification hierarchies,” and so on. I regard modern evolutionary trees as having very little to do with such “logical trees” because evolutionary trees are fundamentally historical diagrams: trees of history. They are representations of a unique, branching sequence of events in the past.
(2) Considered as diagrams of the Natural System, evolutionary trees are much more interesting. The term “the natural system” goes back to the 18th century at least, and refers to the abstract order or pattern that exists in the diversity of life. It is from the idea of the natural system that we get the name of our field, systematics, and also the title of the founding document of the field, Linnaeus’s Systema Naturae (1730s-1760s). Evolutionary trees depict our modern understanding of the natural system, but before Darwin there were many other understandings of what this abstract system was like, and they weren’t necessarily tree-like or historical. In the 19th century the natural system was often represented as a system of interlocking circles, or as stars, or as a map or a net. One of the great pre-Darwinian theorists of the natural system was the British naturalist High Strickland who wondered in 1841 whether the organismal relationships that make up the natural system
With Darwin’s Origin of Species (1859) the natural system becomes the great Tree of Life, and after asking rhetorically
Darwin answers with the only illustration the Origin contains: an evolutionary tree.
In the late 1800s a wide variety of evolutionary trees were published, and the representational devices they employ are diverse, from imaginary cross sections, to timelines, to geography. Interested readers are invited to follow up in two of my papers that are available on the web. (All my links will go to HTML versions, and in most cases these point to pdf versions that include all the figures.)
(3) It is in the context of “trees of history” that evolutionary trees are most interesting, most subtle, and most relevant beyond evolutionary biology. By “trees of history” I mean all manner of genealogical diagrams that depict “descent with modification.” The three principal disciplines that reconstruct trees of history are biological systematics, historical linguistics, and stemmatics (the study of text transmission). For the Darwin-L discussion group that Greg mentioned, I prepared a bibliography on trees of history in all three fields; it hasn’t been updated for some time but I think it’s still a very helpful survey for anyone interested in this issue in a comparative context:
The connections between evolutionary trees and trees of language history were noted in the 19th century, and comparisons between the new historical science of geology and the emerging field of comparative philology were made even before Darwin’s Origin. William Whewell coined the term “palaetiology” in 1837 to refer to these sciences of historical causation, considered as a group. As an indication of how narrow we are today, I heard a molecular biologist give a talk last year in which he announced that biological evolution was like language evolution – an observation he thought no one had made before!
Historical linguists began drawing “evolutionary” trees of languages in the early 1800s, within a very few years of the first trees of biological evolution. In this very same time interval – the first decades of the 1800s – the earliest manuscript stemmata were also published. A “stemma” is an “evolutionary” tree of manuscript descent, illustrating the relationships of the known copies and attempting to reconstruct the state of the (usually missing) ancestral manuscript. The first stemma, published by Schlyter in 1827, includes an absolute time axis and looks very much like Darwin’s evolutionary tree in the Origin (1859), although this similarity seems to be entirely coincidental.
More details on this history, along with samples of early evolutionary, linguistic, and stemmatic trees may be found in two more of my papers:
Thanks to Ed Johnston for extending this thread.
The independence assumption in statistical analysis (so necessary to finesse all those messy and mathematically intractable covariances that appear in real data) is like shipping and handling–it will get you every time. (Recall all those exercises in mathematical statistics beginning with “i.i.d”!) The superb paper by Susan Holmes makes this point very well. Perhaps cladogram-fitting is a bit like factor analysis in social science, a technique used when there is lots of data but not that many ideas. This contrasts, for example, to Feynman diagrams, which use theory to infer a few specific empirical outcomes that provide an independent test of the theory. Tree-fitting must be done, given the state of explanation in the field, but some of the applications do seem to over-reach, more scientism than science.
Here is the most recent draft of my discussion of the skull diagram, from a chapter on causal arrows and linking lines in Beautiful Evidence:
Edward Tufte wrote: “The issue is the external validity of the tree, not just the internal validity within the discipline. That is how I would evaluate a postmodern lit-crit analysis of something, rather than asking if the postmodern analysis was consistent with the current intellectual practices among postmodernites.”
ET asks how we can judge the external validity of a tree. I think he is hoping to see a diagram that separates data and hypotheses, as when you try to fit a regression line to an X-Y plot of data points. You can see the line (hypothesis), you can see the points (data), and you can judge for yourself if the line is a good fit.
These evolutionary trees swallow up the data they are based on, and make it invisible. You actually see only the final tree, and you should be aware that the tree could be the best of a bad lot. The algorithm chooses from a set of candidate trees, even if none of them fits the data very well.
Seeing only the final tree is like viewing a regression diagram containing just the fitted line, with all the data points removed.
Even without a graphical aid, you might hope to see a numerical summary of the confidence that should be reposed in the tree. If you are a statistician, you will probably be disappointed with what you can currently get. As Susan Holmes remarks in a 2003 paper in Statistical Science (volume 18 page 241),
>If you were thinking of using bootstrap values for this purpose, see the cautions listed by Holmes in her paper.
And in case you were hoping that a scientific consensus was forming to back up the style of tree inference used by Ni, Wang, Hu and Li (the authors of the tree example given in ET’s article), you might want to note a statement of Huelsenbeck et al. in Science, 294:2310 (2001):
How much inferential weight will trees bear? Is it ever wise to use a tree for inference?
We shouldn’t prohibit authors like Ni et al. from presenting their trees reconstructed by whatever method they think best. A good figure caption for them to use would be ‘Speculative reconstruction of X.’
But if they want to use a tree for INFERENCE, then I believe they need a different kind of analysis, one whose absence is indicated by ET’s comments.
In the case of the Challenger disaster, there were bad diagrams and good diagrams (see ET’s Visual Explanations, chapter 2). But on the assumption that Ni et al. have presented an inadequate diagram, what could the corresponding good diagram possibly be? My own suggestion (without understanding any of the taxonomic arguments) is that a composite diagram containing dental measurements could be of interest, with data points representing species. Terms like ‘primitive’ and ‘derived’ are used in the article to describe tooth characters without any further explanation. These terms need to be hooked up with specific data values to get a more persuasive case. It would be particularly helpful for them to Include all SIX of the Teilhardina species in a graphic. (Their phylogenetic tree only includes three Teilhardina spp., perhaps because of missing data).
It’s worth noting that Ni et al. DO want to use the tree for inference. They refer to conclusions from it at several points. In particular, they make T. asiatica be an especially early euprimate, “Because T. asiatica is phylogenetically near the root of the euprimate radiation…” Since the tree does not seem strong enough to show this (ET: “every leaf trembles..”) it might be wise to establish the place of T. asiatica among the early primates using more specific arguments that are more closely tied to the measured data.
To sum up, tree inference must face the following challenges:
There is no measure of uncertainty for trees that is both soundly applied and widely used. (The bootstrap assumes independence of characters. Without it, bootstrap error estimates can be off by a large factor). In the Ni et al. paper the bootstrap was not used.
The parsimony algorithm always produces a final tree regardless of quality. Authors should look for other ways to show that conclusions suggested by their tree are actually correct.
‘Calibration’ of tree inference is difficult because true descent is hardly ever known.
DNA analysis of living primates suggests that the fossil record is far from complete. As few as 7% of all the primate species that have ever existed may be known from fossils, according to Tavare et al.(2002): Nature 416:726-729. Ni et al. should be cautious about generalizing from mostly fossil data if they want to reach conclusions about the diurnality of the common primate ancestor.
I wish I had seen this thread back in January, but somehow I missed it. Anyway, you’ve had some highly expert responses from various people, especially Gregory Mayer and Joe Felsenstein, and there’s no point in my repeating their points in less expert words. Nonetheless I’ve always been much less enthusiastic about maximum parsimony than many of its advocates, so I would just say that on the one hand I think you are right to raise the general questions you do, but, on the other hand, you rather give the impression that these questions are not discussed in the systematics literature, whereas in reality they have been very extensively discussed.
There is one point that seems important to me but which appears to have been overlooked in all the comments. Maximum parsimony yields an unrooted tree, but even though you don’t explicitly draw the root you do draw the tree in such a way that virtually everyone will infer the presence of a root between Purgatorius unio and the common ancestor of Altanius orlovi and Teihardina asiatica. So far as maximum parsimony is concerned, however,the root could be absolutely anywhere, between Microchoerus erinaceus and Necrolemur antiquus, for example. In practice, of course, no one would imagine the root was in the latter position, but in ruling out such an absurd location one is going beyond what maximum parsimony allows. Incidentally, all but the rightmost of the little diagrams at the bottom-right of p. 10 show rooted trees, if anyone is unclear what I’m talking about.
It’s many, many years since I last saw it, and maybe I exaggerate what a wonderful book it is, but I recommend you have a look at Numerical Taxonomy, by P. H. A. Sneath and R. R. Sokal
(Freeman, 1973) (not to be confused with Principles of Numerical Taxonomy, written by the same authors ten years earlier). If I remember rightly, this discusses some of the issues that concern you in a very scholarly way.
Dr Tufte,
There may be value in reading Stephen Jay Gould’s short book: “Full House,” if you have not already.
There is an interesting diagram by Bruce MacFadden on page 66 that illustrates the complex branching bush of Equine evolution. The chapter may also shed some light on the notion of cause and “progress” in this context.
I think you will also enjoy his explanation for the disappearance of the .400 batting average in baseball as a tightening of variation. (see page 119)
Assembled by the KDE gang and lofted to the top of reddit a week and a day after the release of Debian Etch (see diagram for Debian’s lineage), a cladogram of on of the cybergenic kingdoms of the Internet. Click for a closer view. The evolution of code.
What is the significance of the position of the labelled bullets on the timeline? Is there not a danger that, with a constant 45¿¿ branching angle, the position of the *branch point* might be mistakenly assigned that significant meaning? Both points can’t be meaningful, given the way the versions are given each their own position in vertical space based only on the need to stack them on top of each other.
Derek,
I posted your question on the image’s thread for the artist.
Version 7.5 of the Linux tree has been published (see link in Niels’ post and click the download link half way down the page) with a slightly modified – and more strict – layout. For instance, the angled branches were remodeled, and the GNU/Linux icon was removed;
the kernel itself came about in 1991 but the name GNU/Linux was not used until far later. All the additions has made the image larger in height than width, and perhaps it would look better if it was redesigned vertically.
Is cladogram a better definition for this image, even though it is also a timeline?
Well, there’s little doubt but that it is a cladogram in-so-far as it is a drawing of branches, and it adheres remarkably well to the Wikipedia definition of cladistics. That you have added a strict interpretation of time only adds to the virtue of the diagram. That’s something biologists wish they had. From a design perspective, I think you could do away with the big dots at corners and reserve dots for new origins (looking closely there is clearly more than origin). This would make origins more visually distinctive, which they ought to be.
I think it would actually be very useful as a classroom example because it nicely illustrates several evolutionary phenomena in one real-world example. Founder effect, convergence, extinction. I’m not sure we can demonstrate a Hardy-Weinberg equilibrium, but maybe if you added in the developers names . . .
What is the purpose of the color scheme? Do varous shades of blue indicate, say, age of latest version or all those derived from Debian? Does a dark color indicate the number of RPMs in repositories? Is it a map-like problem that you’re trying to pick colors that adequately separate lines visually?
Excellent, thanks for clearing that out. I might stick to calling it a timeline though, since the word makes more sense. I didn’t know what a cladogram was until I found this site. But maybe that’s just me. 🙂
Currently the dots do not only mark the origin but also changes to the name name or other fundamentals. In many cases because companies acquired the projects – or the opposite, companies go down and the project lives on with another name. I think I’ll swap those for a short vertical marker.
I don’t think removing the dots at corners as you suggested would be consistent.
Also, so far I’ve tried to maintain a strict layout rather than adding explanatory texts all over. It would take up too much space.
> “or all those derived from Debian?”
No, the colors is just cosmetics, the distributions connected to Debian are derived from Debian.
Thanks for the input so far! I really appreciate it.
This discussion reminds me of some recent reading I did on horizontal gene transfer and Christopher Alexander’s essay A City is not a Tree — namely around the consideration of semilattices over trees as a fundamental structure, and the notion that people fundamentally have trouble with interlinked and overlapping structures. I am no geneticist (or mathematician, or city planner, for that matter), but it’s interesting to think about.
I thought this was an interesting article, if only for reminding me of Darwin’s initial sketches.
It seems to me that the issue is that while the tree is large, and while we can imagine mavigating through a 3D or even 4D space with some digital tools, the whole tree rarely if ever needs visualization…. even with all those fancy tools. No substantive question could be addressed without considerable and theoretically justified discounting (though not necessarily censoring) of uninteresting branches.
Indeed while the imagery of trees and branches may be compelling from an historical and simplistic point of view, the notion that genes combine, and recombine seems to be more suggestive of a massive banyan tree/vine, parts wrapping unintelligibly around a base, but where branch, tree and vine are a complex tangle, constantly diverging, constantly recombining.
Perhaps the problem lies first and foremost in the wrong metaphor in a simple “linear” tree… genetic evolution is certainly linear, but not unremitingly so, and attempts to draw it like this are finally failing under their own success. Sounds like a good time to be a quantitative geneticist with an interest in visual display of data.
Carl Zimmer, “Crunching the Data for the Tree of Life” New York Times, February 10, 2009 here
The issue here is too much data and not enough ideas. Finer and finer distinctions fitted by more and more credulous models generate an enormous pile of nouns and adjectives rather than the verbs of explanation. In addition, most large data sets have big redundancies, colinearities, non-independent observations, serial correlation, and bang-bang duplicates–and so the effective N is much smaller than the nominal N.
Here’s a 1934 tree that includes time (roughly measured) in it’s tree:
Source: Constance Areson Clark, “Evolution for John Doe: Pictures, the Public, and the Scopes Trial Debate,” The Journal of American History, 87 (March 2001), 1275-1303.
Constance Areson Clark’s paper is an excellent history of the evolutionary diagrams surfacing in the notorious Scopes Trial. Her paper is here.
Dear E.T.,
You write:
Reasoning about tree diagrams evokes not only specifics about the content at hand but also more general issues about analytical credibility. What causal mechanisms are assumed or described by the tree? In what space does the tree reside? What does the data matrix behind the tree look like? Can we see that matrix? What is the interplay of evidence and assumption in generating the results? Do alternative trees fit the data? How many? Can the published tree be distinguished from alternatives? Does the tree reflect good practice in reporting evidence?
Couldn’t all these questions be asked about the tree-like diagram about the history of marketing trends and stylistic patterns in the development of pop/rock music by Reebee Garofalo and Damon Rarey? Is the difference that the work by Reebee Garofalo is not published as part of a scientific publication? On the other hand, the chart by Garofalo and Rarey has a y axis and at least pretends to graph record volume sales (however, the hand lettering, the very smooth curves and the almost constant distance between curves suggests that we shouldn’t take those volume sales serious at all; but then: how good is the graphing of data?).
Am I right to assume that you apply very different standards to the chart by Garofalo and Rarey than to cladograms published in the scientific literature?
ET answer: yes.
Evolution as confection
While the edited (and printed) version of these pages have been much improved from the original draft shown thanks to the wonderful insight of Greg, Bob, Joe, Zen, and others, there is a notable error I felt I should point out, although it may be a bit fussy.
On the top of second page in the revised image (page 75 in BE), ET says, “Thus names closer to one another have more in common than names further apart”. On the average, this is true, but again, relationships are indicated by the branching pattern, not Y-space adjacency. Species that share a more recent node are more closely related. Organisms that branch from the terminal nodes are next to each other and more closely related to each other than any are to any other species.
However, adjacency is not always indicative of close relations. Look at Teilhardina asiatica and Teilhardina americana, for example. Despite the fact they are very close together in space, the branching pattern indicates (by my count) 5 nodes between them,
which is quite distant. As Zen Faulkes has already noted, this indicates that T. americana is actually likely a member of a different genus, and we should rethink our taxonomic classification of this group.
As further examples, Tarsius sp. and Loveina zephyri, Teilhardina asiatica and Galagoides demidoff, Donrusselia sp. and Altanus orlovi, and Plesiolestes problematicus and Purgatorius unio all provide examples of species located adjacently in Y-dimensional space that are actually quite distantly related.
I propose a simple change that would account for this: “Thus names closer to one another often have more in common than names further apart”. More precisely, you could say “Thus names that share a recent branching point have more in common than names that share a more distant branching point”.